- Products
- CONFLEX DOCK Algorithm
CONFLEX DOCK is a program for protein-peptide docking based on a four-body statistical coarse-grained potential.
Overview of the Four-Body Statistical Coarse-Grained Potential
Using a coarse-grained model with representative points for amino acid residues of proteins, the stabilities of native and non-native forms are compared. The potential score is calculated from a tetrahedral representation obtained by Delaunay triangulation of the representative points.
Krishnamoorthy, B.; Tropsha, A. Bioinformatics, 19, 1540–1548 (2003).
This four-body statistical coarse-graining was applied to protein-peptide docking.
T. Aita, K. Nishigaki, Y. Husimi, Comput. Bio. Chem. 34, 53–62 (2010).
Based on this four-body coarse-grained potential, the protein-peptide docking program was developed.
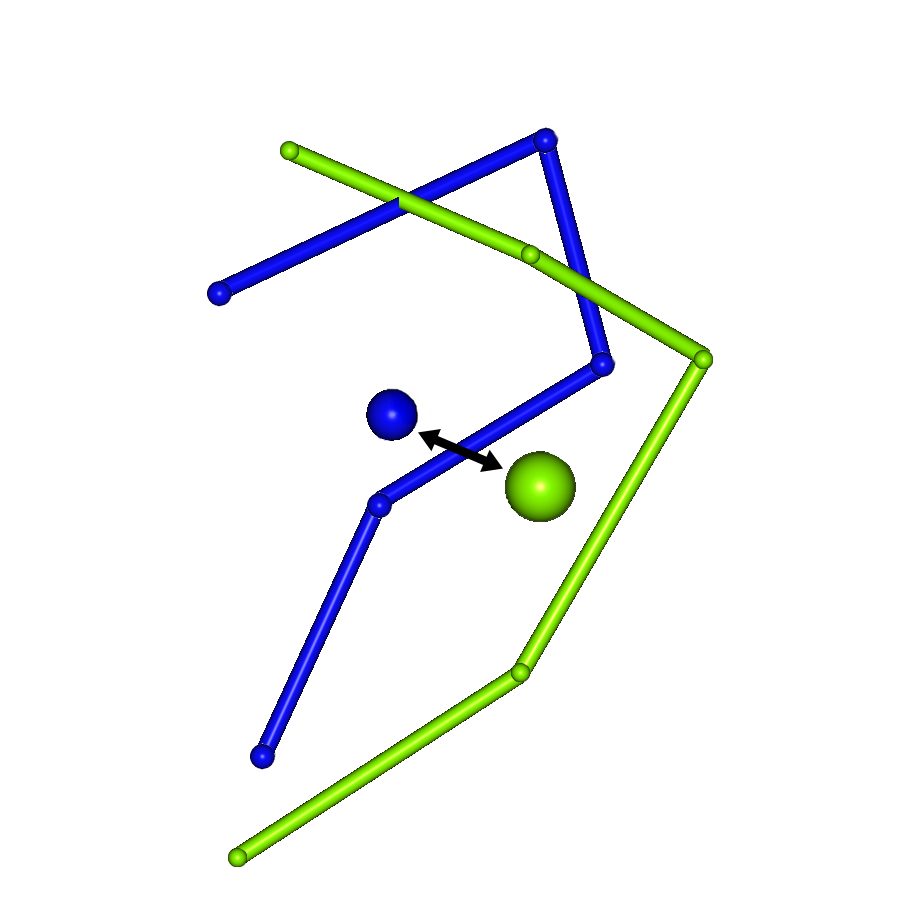
Combinations of Four Amino Acid Residues
To calculate the potential score, tetrahedra consisting of four amino acid residues are considered. Each tetrahedron belongs to one of the following five classes:

| Class | α | Description |
|---|---|---|
| {1,1,1,1} | 0 | All four residues are non-consecutive in the primary sequence |
| {2,1,1} | 1 | One pair of consecutive residues, the other two are non-consecutive |
| {2,2} | 2 | Two pairs of consecutive residues, each pair is non-consecutive with the other |
| {3,1} | 3 | Three consecutive residues, and one non-consecutive residue |
| {4} | 4 | All four residues are consecutive in the protein’s primary sequence |
Potential Score
The potential score is calculated using the following formula:
Here, is the observed frequency and is the expected frequency.
The observed frequency is obtained by dividing the protein via Delaunay triangulation to create many tetrahedra, and then counting them. The division into tetrahedra is performed so that the minimum angle is maximized and the circumscribed sphere is minimized.
The expected value P is calculated by the following formula:
- , , , : Occurrence frequency of residues i, j, k, l in the dataset
- : Probability that the tetrahedron class is α
- η: Number of types of amino acid residues composing the tetrahedron
- tν: Number of residues of type ν in the tetrahedron
Simulation Procedure of CONFLEX DOCK
Docking simulation with CONFLEX DOCK proceeds as follows:
- Place search points on the protein surface
- Explore peptide positions (binding poses)
- Sort results based on potential score and prediction accuracy indices (RMSD, GTGD)
- Cluster binding poses
Placement of Search Points - Defpol Method
Search points are placed spherically to cover the protein, and then moved to the surface toward the protein centroid.
Pomelli, C. S.; Tomasi, J.
J. Comput. Chem., 1988, 15, 1758–1776.
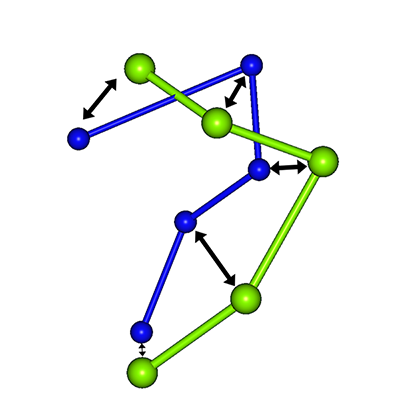
Search Algorithm
The docking search proceeds as follows:
- Place a peptide amino acid residue on a search point; form a tetrahedron with three protein residues and evaluate the score
- Place the next peptide residue on another search point and evaluate the score
- Select search points based on predetermined criteria
- Repeat the above evaluation and search process
This procedure is treated as a tree structure. An elite strategy is used, where N nodes with the highest scores are selected for the next search. The figure below shows the case where N = 2.

Evaluation Methods for Prediction Accuracy
RMSD: Root Mean Square Deviation
A method to compare the positions of each amino acid residue.
- N: Number of peptide amino acid residues
- ri: Coordinate of the i-th amino acid residue point
- riexp: Coordinate of the i-th residue point in the experimental structure
GTGD: Geometric center To Geometric center Distance
An index representing the degree of coincidence of geometric centers.
- N: Number of peptide amino acid residues
- ri: Coordinate of the i-th amino acid residue point
- riexp: Coordinate of the i-th residue point in the experimental structure