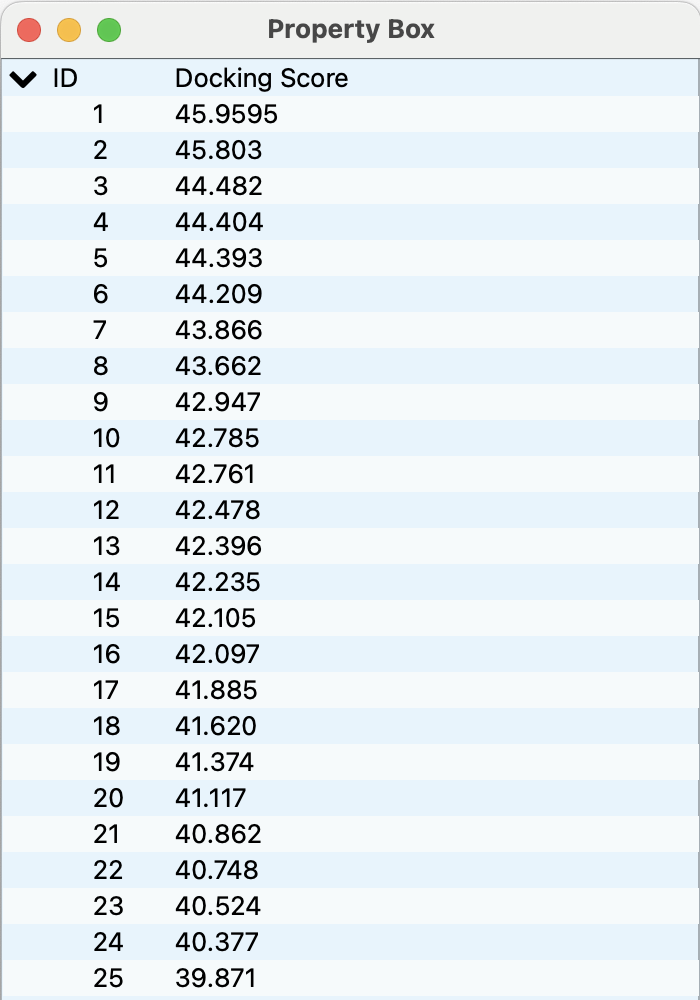
Protein–peptide docking simulation
CONFLEX DOCK is a docking simulation program that predicts where a specified peptide chain will bind to a protein and form a complex.
This section explains how to create input files for the simulation, execute the calculation, and display the results. Here, Krev interaction trapped protein 1 (PDB ID: 4hdq) is used as the protein.

Input files
Using 4hdq.pdb downloaded from the Protein Data Bank, prepare the input data required for the calculation.
First, extract the coordinate data for chain A (lines 596 to 3138) and save it as a separate file named “4hdq_a.pdb”.
Contents of 4hdq_a.pdb
ATOM 1 N TYR A 419 10.722 15.896 36.775 1.00 65.31 N ATOM 2 CA TYR A 419 11.521 15.928 35.512 1.00 67.80 C ATOM 3 C TYR A 419 11.189 17.152 34.645 1.00 55.21 C ATOM 4 O TYR A 419 10.052 17.636 34.579 1.00 56.45 O ATOM 5 CB TYR A 419 11.346 14.624 34.690 1.00 75.21 C ... ... ATOM 2540 CG LEU A 729 31.167 9.539 -1.671 1.00 47.66 C ATOM 2541 CD1 LEU A 729 30.013 8.839 -0.939 1.00 44.71 C ATOM 2542 CD2 LEU A 729 30.768 10.892 -2.272 1.00 51.04 C TER 2543 LEU A 729
Next, specify the peptide sequence to be docked.
The peptide sequence can be specified either by entering a one-dimensional sequence or by preparing and loading a PDB file.
Since 4hdq.pdb contains chain C, a 5-residue peptide (ARG-ARG-ASP-TYR-PHE, lines 4440 to 4481), this data will be used as input.
Save it as a separate file named “4hdq_peptide.pdb” in the same directory as 4hdq_a.pdb.
Contents of 4hdq_peptide.pdb
ATOM 3845 N ARG C1377 30.216 23.590 21.799 1.00 57.06 N ATOM 3846 CA ARG C1377 29.684 24.981 21.860 1.00 54.44 C ATOM 3847 C ARG C1377 29.284 25.473 20.457 1.00 59.72 C ATOM 3848 O ARG C1377 28.148 25.942 20.266 1.00 56.63 O ATOM 3849 CB ARG C1377 30.678 25.967 22.520 1.00 53.04 C ... ... ATOM 3883 CE1 PHE C1381 25.487 22.366 10.239 1.00 29.96 C ATOM 3884 CE2 PHE C1381 24.065 20.653 9.391 1.00 26.70 C ATOM 3885 CZ PHE C1381 24.252 21.944 9.721 1.00 28.89 C TER 3886 PHE C1381
Docking calculation
Execution from Interface
Open “4hdq_a.pdb” in the CONFLEX Interface, select “Docking” from the Calculation menu, and set the calculation conditions.
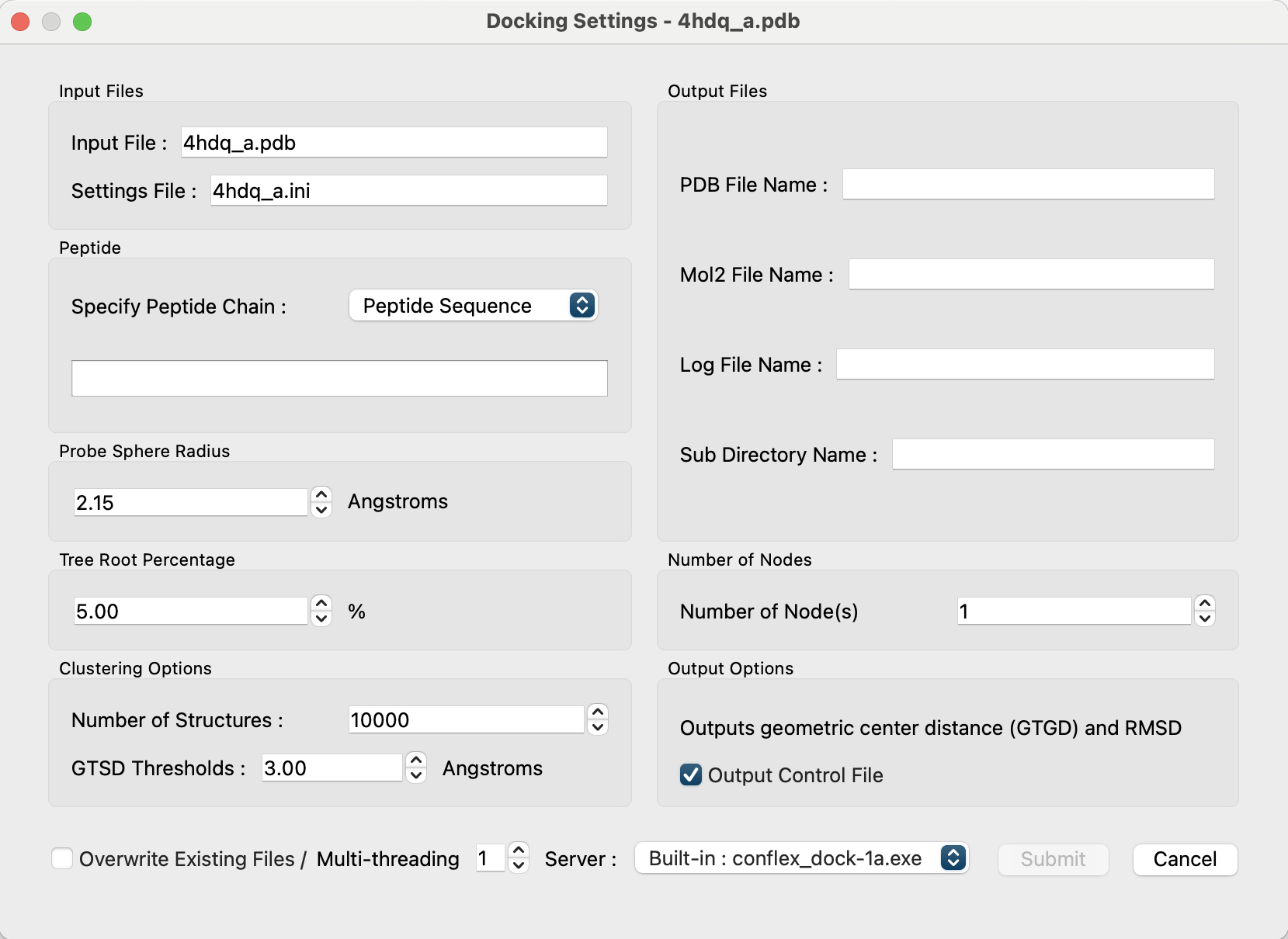
First, set “Specify Peptide Chain:” in the “Peptide” section to [Open File].
Then, the button will appear below it.
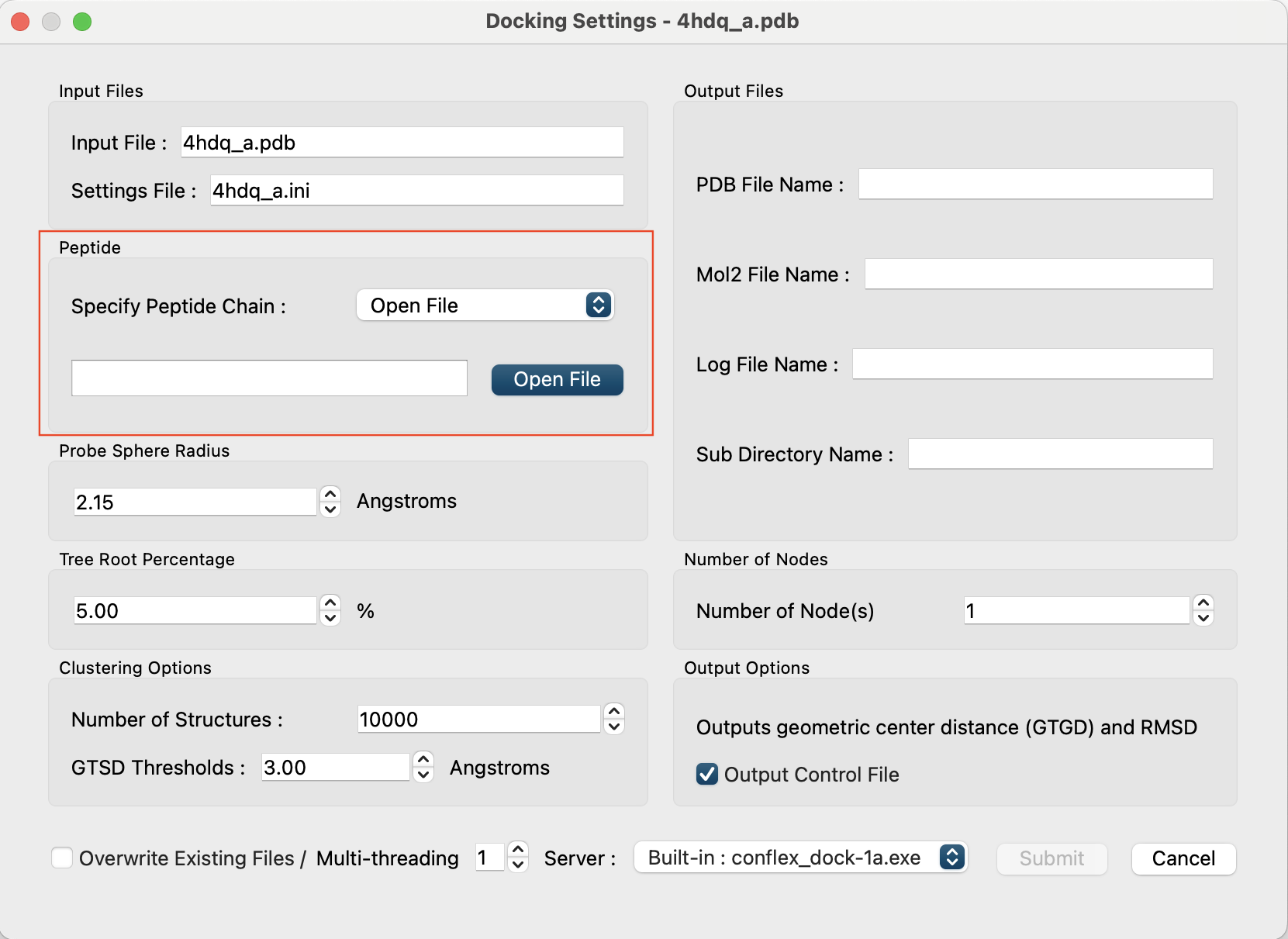
Click and select the “4hdq_peptide.pdb” file you created.
After loading the file, the file name and the sequence of residues will be displayed, and in the “Output Files” section, the output file name and directory name will be automatically set as a combination of the input PDB file name and the peptide's amino acid sequence (for example, “4hdq_a_ARG-ARG-ASP-TYR-PHE”).
Each of these names can be changed individually. If you wish to change them, please edit the boxes directly.
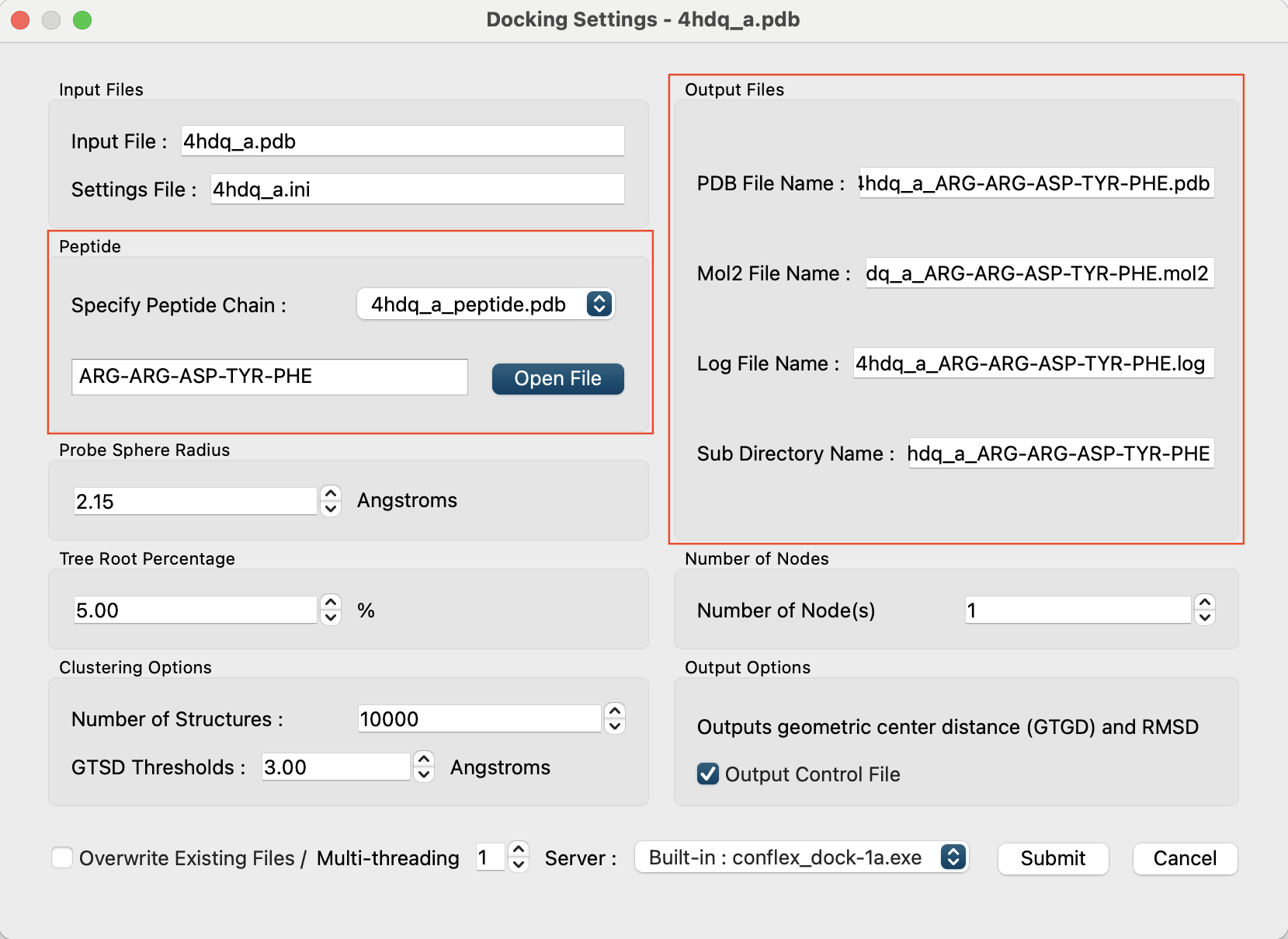
Click the button at the bottom right of the dialog to start the calculation.
The Job Manager will then launch, displaying “docking” in the “Program” column, the peptide's amino acid sequence in the “Job Type” column, and the protein PDB file name in the “Molecule” column.
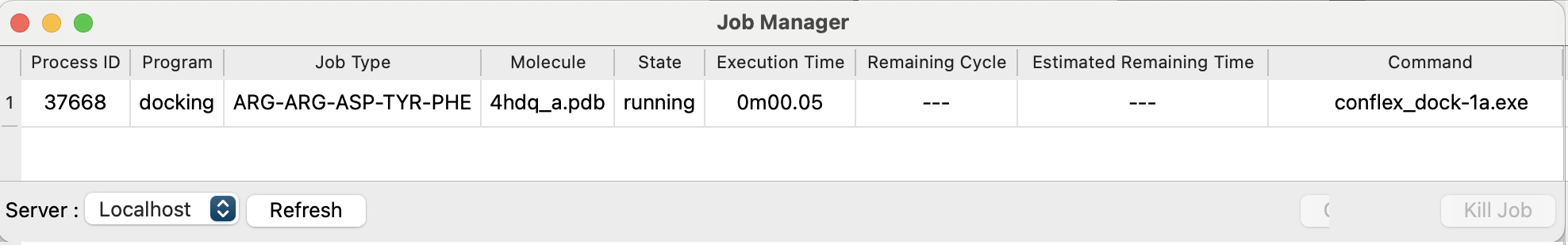
After the “Status” column changes from “running” to “Finished”, double-click the job row to open one of the output files, “4hdq_a_ARG-ARG-ASP-TYR-PHE.pdb”.
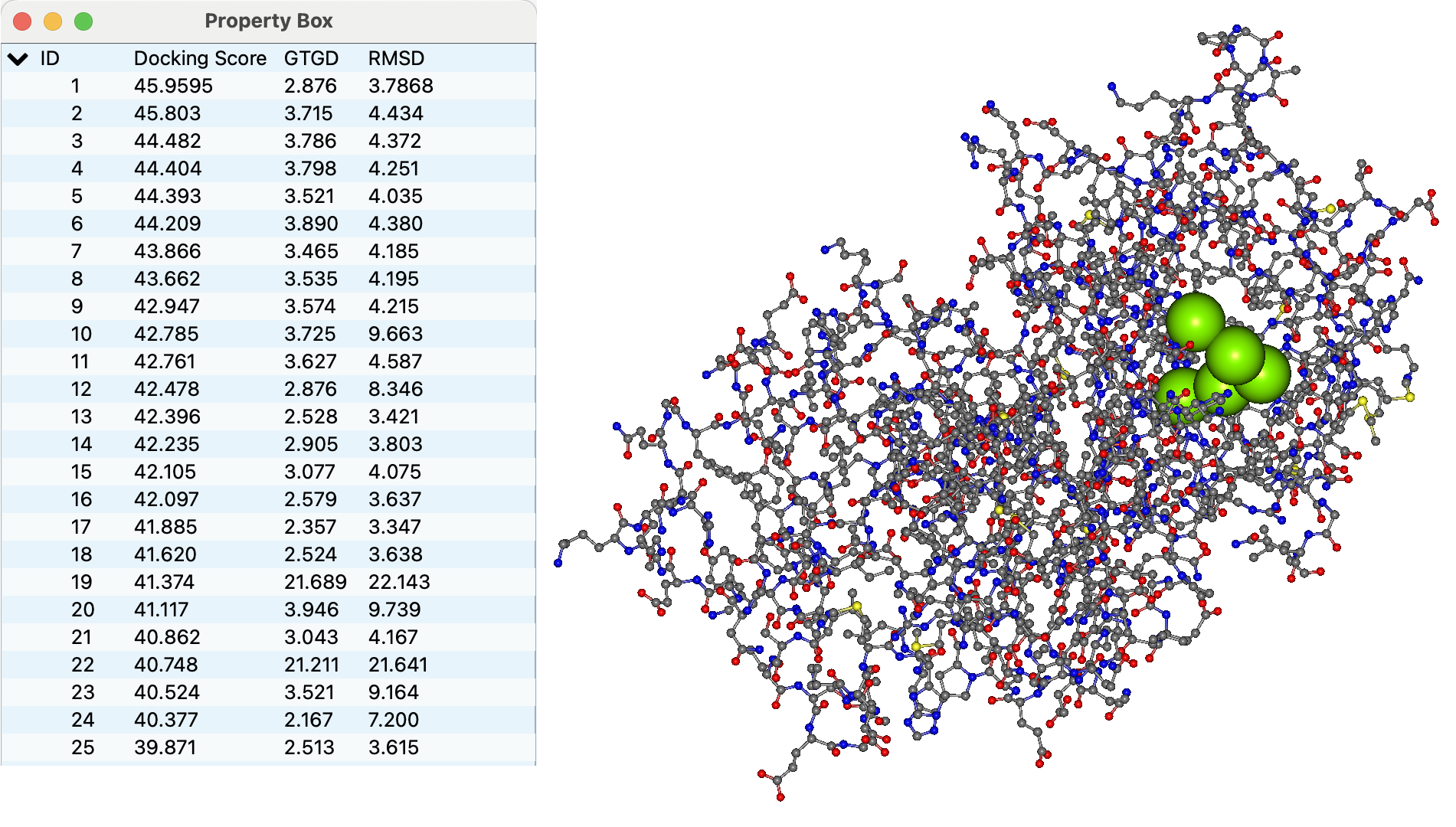
The representative points of the peptide's amino acid residues in each docking pose are shown as connected green spheres.
In the Property Box, the Docking Score, the Geometric center To Geometric center Distance (GTGD, Å), and the Root Mean Squared Deviation (RMSD, Å) for each pose are displayed.
Clicking each ID row in the Property Box switches the displayed docking pose.
Output files
When the calculation is complete, a “4hdq_a.ini” file reflecting the settings and the following three output files:
- 4hdq_a_ARG-ARG-ASP-TYR-PHE.log
- 4hdq_a_ARG-ARG-ASP-TYR-PHE.mol2
- 4hdq_a_ARG-ARG-ASP-TYR-PHE.pdb
Additionally, a directory named “4hdq_a_ARG-ARG-ASP-TYR-PHE” is created and the files are saved there.
The “4hdq_a.ini” file is saved with the following contents.
probe_rad=2.15 root=0.05 branch=1 cluster=10000 cluster_dist=3
“4hdq_a_ARG-ARG-ASP-TYR-PHE.log” contains the progress of the calculation, score values and distributions for each docking pose, clustering results, and more.
==================================================================================================================================
_/_/_/_/ _/_/_/_/ _/ _/ _/_/_/_/ _/ _/_/_/_/ _/_/ _/_/ _/_/_/ _/_/_/_/ _/_/_/_/ _/ _/_/
_/ _/ _/ _/_/ _/ _/ _/ _/ _/ _/ _/ _/ _/ _/ _/ _/ _/
_/ _/ _/ _/ _/ _/ _/_/_/_/ _/ _/_/_/_/ _/ _/ _/ _/ _/ _/ _/_/
_/ _/ _/ _/ _/_/ _/ _/ _/ _/ _/ _/ _/ _/ _/ _/ _/ _/
_/_/_/_/ _/_/_/_/ _/ _/ _/ _/_/_/_/ _/_/_/_/ _/_/ _/_/ _/_/_/_/ _/_/_/_/ _/_/_/_/ _/ _/_/
Start --- CONFLEX DOCK ver.1.A.0303 (Updated March, 3rd, 2025)
Date: 2025/04/15
Time: 00:23:29
==================================================================================================================================
<< Licensee information >>
Licensee name: CONFLEX DOCK User
Licensing institution: CONFLEX Corporation
Address: Shinagawa Center Bldg. 6F, 3-23-17 Takanawa
City: Minato-ku, Tokyo
ZIP code: 108-0074
Nation: Japan
Telephone number: +81-3-6380-8290
E-mail address: info@conflex.co.jp
Matched MAC address: 11:22:33:44:55:66
DATE OF THE LICENSE START: 2025/04/14
DATE OF THE LICENSE EXPIRE: 2026/12/31
==================================================================================================================================
Maximum number of threads: 8
Current number of threads: 1
Directory containing Potential and License files: /Applications/CONFLEX/par
Representative point of residue: C_alpha atom
==================================================================================================================================
*** Delaunary tessellation of protein and setting of search points
Probe radius (Angstrom): 2.150
Protein file name: 4hdq_a.pdb
Number of amino acid residues in the protein: 311
Peptide file name: 4hdq_a_peptide.pdb
Number of amino acid residues in the peptide: 5
Score of peptide on the nearest search points: 30.867
GTGD of peptide on the nearest search points from input structure (Angstrom): 0.653
RMSD of peptide on the nearest search points from input structure (Angstrom): 1.562
CPU time (s): 0.734
Wall time (s): 0.734
==================================================================================================================================
“4hdq_a_ARG-ARG-ASP-TYR-PHE.mol2” contains each docking pose, with the amino acid residues of the protein also represented as representative points.
The following ten files are output in the “4hdq_a_ARG-ARG-ASP-TYR-PHE” directory.
4hdq_a_4hdq_a_peptide_ClustPep_N1_DIST3.csv 4hdq_a_4hdq_a_peptide_FRank_N1_0.csv 4hdq_a_4hdq_a_peptide_ClustSite_N1_DIST3.csv 4hdq_a_4hdq_a_peptide_GRank_N1_0.csv 4hdq_a_4hdq_a_peptide_Clust_N1_DIST3.mol2 4hdq_a_4hdq_a_peptide_N1_ALL.csv 4hdq_a_4hdq_a_peptide_Clust_N1_DIST3.pdb 4hdq_a_4hdq_a_peptide_RRank_N1_0.csv 4hdq_a_4hdq_a_peptide_Distribution_N1.csv searchpoint.mol2
Among these, the contents of the .csv files are as follows.
| File name | Description |
|---|---|
| 4hdq_a_4hdq_a_peptide_ClustPep_N1_DIST3.csv | Clustering number (CNo), Score rank (FRank), Score (FSum), Calculation order (RNo), Line number (LNo), GTGD、RMSD |
| 4hdq_a_4hdq_a_peptide_ClustSite_N1_DIST3.csv | Clustering number (CNo), Number of poses in each cluster (Nc), Score average (weighted FAve(weighted), Unweighted FAve(noweighted)), Score variance (unweighted average FVar), Maximum score (FMax), Minimum score (FMin) |
| 4hdq_a_4hdq_a_peptide_Distribution_N1.csv | Score distribution |
| 4hdq_a_4hdq_a_peptide_FRank_N1.csv | Score rank (FRank), Score (FSum), GTGD rank (GRank), GTGD, RMSD rank (RRank), RMSD, Calculation order (RNo), Line number (LNo) |
| 4hdq_a_4hdq_a_peptide_GRank_N1.csv | GTGD rank (GRank), GTGD, RMSD rank (RRank), RMSD, Score rank (FRank), Score (FSum), Calculation order (RNo), Line number (LNo) |
| 4hdq_a_4hdq_a_peptide_N1_ALL.csv | Calculation order (RNo), Line number (LNo), Position points of each residue, Score (FSum) |
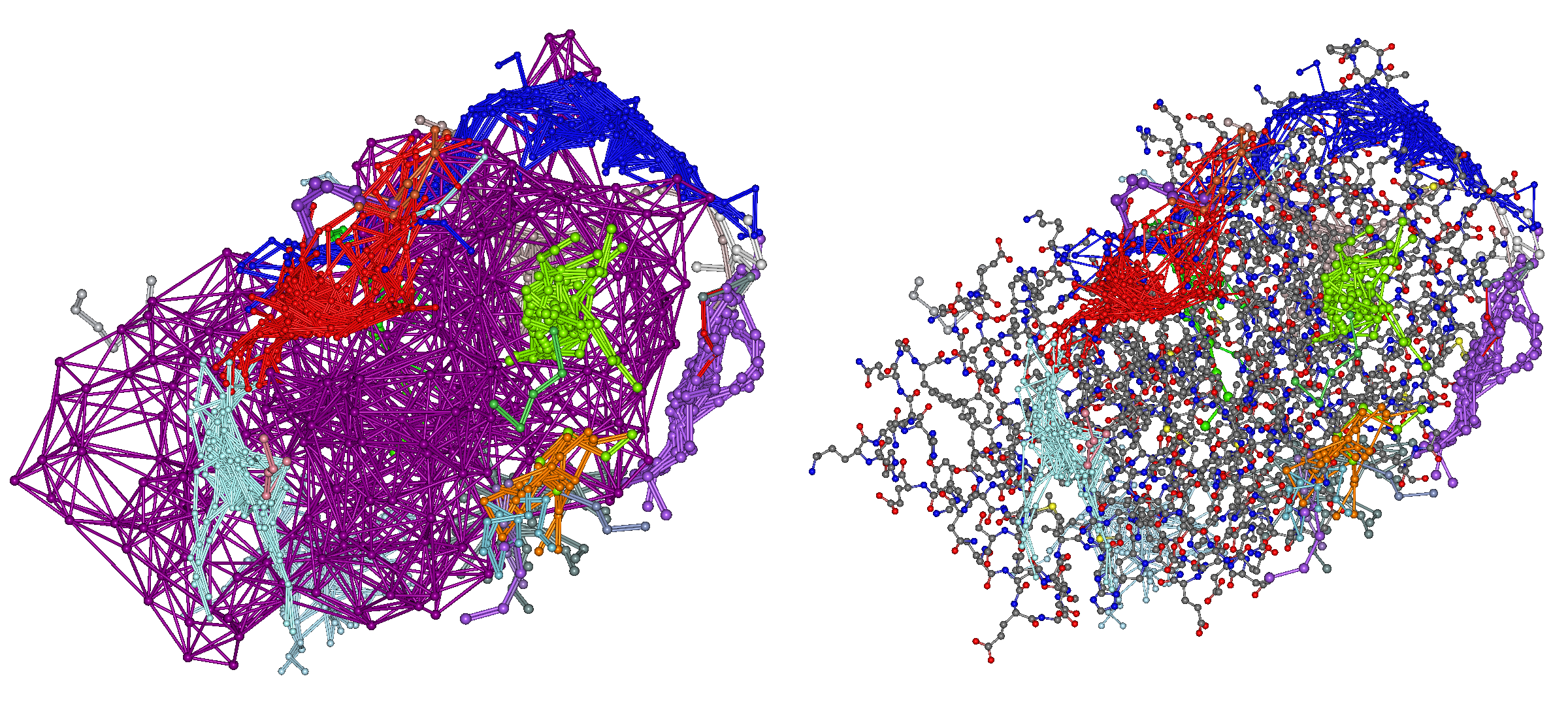
All docking poses are output in clustered form in “4hdq_a_4hdq_a_peptide.mol2” (left) and “4hdq_a_4hdq_a_peptide.pdb” (right), and are displayed as shown below.
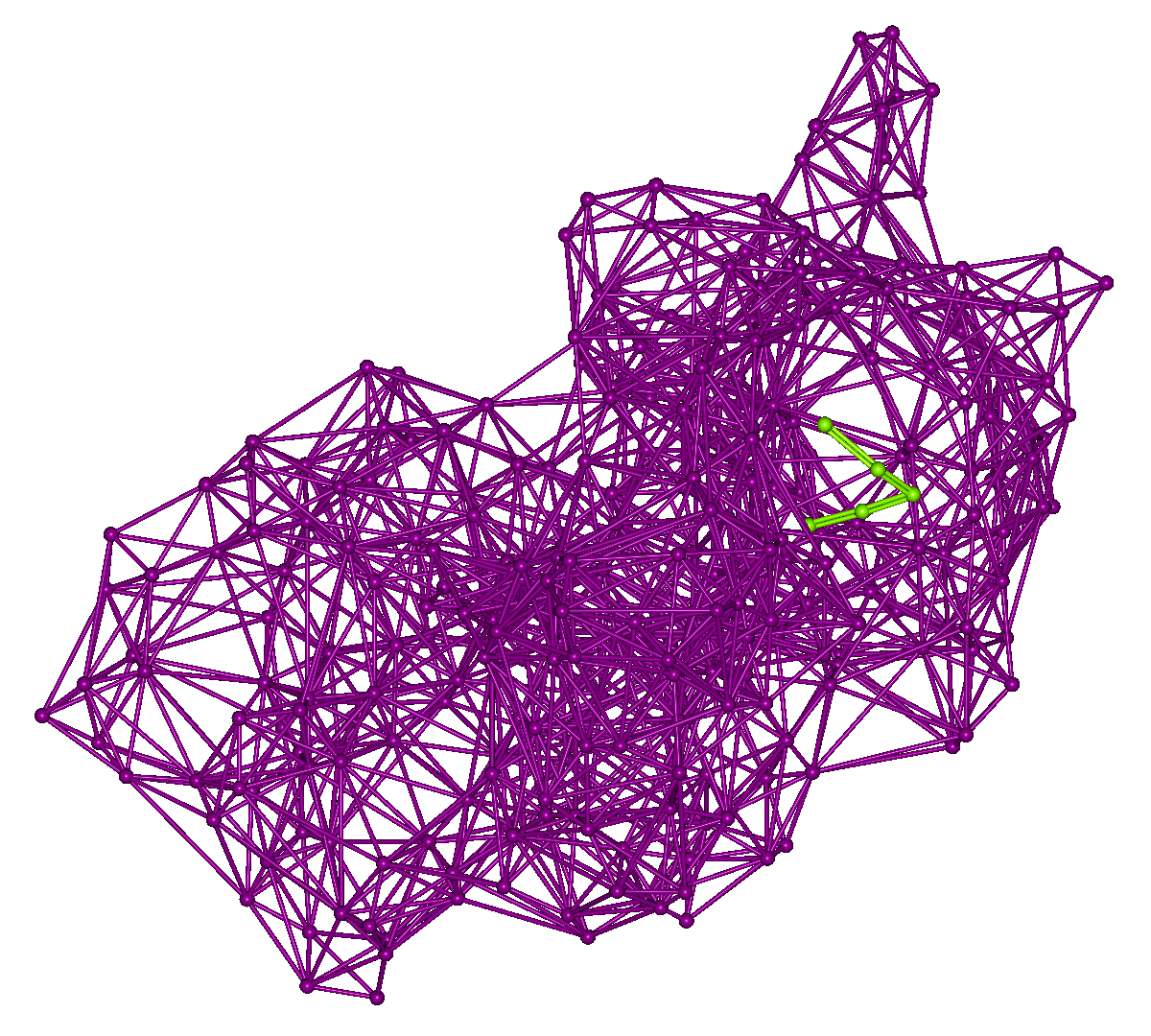
In “search.mol2,” the representative points of the protein's amino acid residues and the search points for the peptide placed on its surface are output.

Specifying a peptide sequence
It is also possible to directly enter the amino acid sequence of the peptide to be searched in the “Peptide” field of the Docking Settings screen and perform the calculation.
If the sequence is the same, the docking score results will not differ from those obtained by specifying a PDB file.
The sequence can be specified using either the one-letter code (left) or the three-letter code (right). However, when entering the three-letter code, please insert a hyphen (“-”) between each residue.
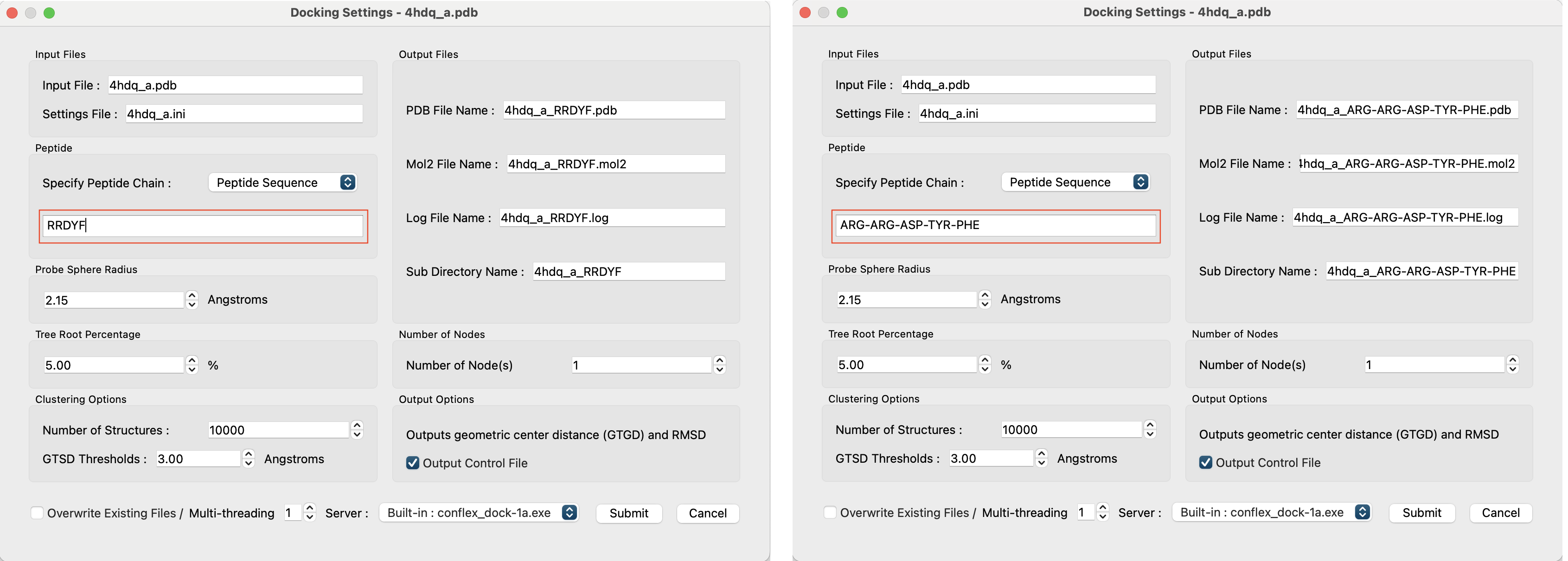
However, since there is no reference structural data, GTGD and RMSD will not be output as shown below.